| INNOV.RU | Информационный портал |
Методика проведения компьютерного моделирования процесса ионной имплантации методом молекулярной динамики с помощью пакета программ для классической молекулярной динамики LAMMPS
The technique of computer simulation of ion implantation using LAMMPS
УДК 004.942
Выходные сведения: Сундукова О.А., Белова И.М. Методика проведения компьютерного моделирования процесса ионной имплантации методом молекулярной динамики с помощью пакета программ для классической молекулярной динамики LAMMPS // Иннов: электронный научный журнал, 2017. №3 (32). URL: http://www.innov.ru/science/tech/metodika-provedeniya-kompyuternogo-/
Авторы:
Сундукова О.А.1, Белова И.М.2
1 магистрантка, ФГБОУ ВО Московский политехнический университет, г. Москва, Российская федерация (107023 г. Москва, ул. Большая Семеновская, д.38), e-mail: soabox12@gmail.com
2 к.ф.-м.н., доцент кафедры «Информатика и вычислительная техника», ФГБОУ ВО Московский политехнический университет, г. Москва, Российская федерация (107023 г. Москва, ул. Большая Семеновская, д.38), e-mail: irbelova1@gmail.com
Authors:
Sundukova O.A. 1, Belova I.M.2
1 master, Moscow Polytechnic University, Moscow, Russian Federation (Bolshaya Semenovskaya str., 38, Moscow, 107023.), e-mail: soabox12@gmail.com
2 Ph.D., assistant professor of dept. “Informatics and Computer Engineering”, Moscow Polytechnic University, Moscow, Russian Federation (Bolshaya Semenovskaya str., 38, Moscow, 107023.), e-mail: irbelova1@gmail.com
Ключевые слова: ионная имплантация, метод молекулярной динамики, LAMMPS
Keyword: ion implantation, molecular dynamics method, LAMMPS
Annotation: Article is considering process of software preparation for molecular dynamics modelling with LAMMPS and modelling ion implantation for Cu-Fe ion-target combination. Description of setting the environment, configuration of input file, algorithm for processing results are presented. Graphs of the admixture distribution profile and the distribution of the point defects along the depth of the target are presented. An analysis of the presence of defects in the crystal structure was carried out using the construction of the Voronov diagram. Modelling results were processed using a Python script. Based on the results of the simulation, it was concluded that in order to carry out computer simulation of the ion implantation process using the package for classical molecular dynamics of LAMMPS, it is necessary to select the equipment based on the expected volumes of the task, adjust the software and generate an input file taking into account the elements participating in the simulation. On the basis of the results obtained, one can expect that: the greater the energy of the implanted atoms, the deeper the admixture and point defects are located; the maxima of the distribution of point defects will be deeper than the maxima of the admixture distribution.

Введение
Для моделирования процесса ионной имплантации используется несколько различных программных пакетов.
В работах [8], [9], [10] используется пакет SRIM. Он имеет ряд достоинств, таких как: удобный пользовательский интерфейс, встроенные параметры по умолчанию для известных науке ионов и материалов, возможность рассчитывать электронную тормозную способность любого иона в любом материале на основе усреднения параметров широкого спектра экспериментальных данных. Тем не менее, при расчетах не учитывается ни кристаллическая структура, ни динамические изменения состава материала, и это серьезно ограничивает полезность пакета в некоторых случаях. Кроме того, исходный код проекта не доступен для пользователя. Главной отличительной чертой программы SRIM (TRIM) является использование для расчета угла рассеяния так называемой “магической” формулы. Магическая формула может быть использована только для весьма ограниченного числа потенциалов ион-атомного взаимодействия, что существенно ограничивает ее использование для решения ряда важных прикладных задач [6].
В работе [11] используется GEANT4 — инструментарий для моделирования прохождения элементарных частиц через вещество с использованием методов Монте-Карло [5]. В отличие от SRIM, GEANT4 обладает открытым исходным кодом, что позволяет исследователю вносить свои правки в исходный код, переопределяющий ключевые моменты симуляции. Серьезным недостатком этого пакета является то, что он не позволяет воссоздать точную картину образовавшихся в процессе ионной имплантации дефектов. Он дает возможность определить, что дефекты в мишени имеются по потерям энергии, но как именно они образовались и как выглядят - нет.
В работе [12] используется LAMMPS — свободный пакет программ для классической молекулярной динамики, разработанный̆ группой из Сандийских Национальных Лабораторий. Разработчики пакета учли возможность применения его при поведении масштабных расчётов (до десятков миллионов атомов). Пакет распространяется по лицензии GPL и доступен в виде исходных кодов.
Основные особенности программного пакета LAMMPS [4]:
- поддержка большинства двухчастичных и многочастичных короткодействующих потенциалов межатомного взаимодействия;
- в системах, в которых есть кулоновское взаимодействие, для расчета сил могут быть использованы реализованные методы Эвальда и PPPM;
- при расчетах короткодействующих сил нужно учесть всех соседей частицы. Для этого в LAMMPS применяются списки;
- на многопроцессорных системах производится пространственная декомпозиция расчетов;
- несмотря на то, что LAMMPS используется для больших расчетов и поэтому по умолчанию компилируется в параллельной версии, его можно настроить и для запуска в однопроцессорном режиме;
- выгрузку атомных конфигураций можно производить как в текстовый, так и в бинарный файлы. Начальная же конфигурация атомов для расчета может быть как сгенерирована в программе, так и прочитана из бинарного/текстового файла;
- присутствуют возможности анализа атомной конфигурации «на лету»;
- параллельные расчеты можно проводить не только с использованием MPI, но и технологию CUDA. К сожалению, графические процессоры можно использовать только для потенциалов Леннард-Джонса и Кулона.
LAMMPS позволяет решать уравнения движения Ньютона для коллекций атомов, молекул или макроскопических частиц, которые взаимодействуют с помощью сил кратко- или долгосрочной дальности с различными начальными и/или граничными условиями [14]. Для вычислительной эффективности LAMMPS использует список соседей, чтобы сохранять треки близлежащих частиц. Списки оптимизированы для систем с частицами, которые отталкиваются на коротких расстояниях, так что локальная плотность частиц никогда не становится слишком большой. На параллельных машинах, LAMMPS использует методы пространственного разложения для разделения домена моделирования на небольшие 3d поддомены, каждый из которых назначается отдельному процессору. Процессоры общаются и хранят информацию об атомах, содержащихся в их поддомене для атомов, которые находятся в соседних поддоменах. LAMMPS является наиболее эффективным (в параллельном смысле) для систем, частицы которых заполняют 3d параллелепипед с примерно одинаковой плотностью.
Наиболее перспективным и удобным для моделирования процесса ионной имплантации в металлических мишенях представляется пакет LAMMPS. Он позволит не только смоделировать распределение примеси и рассчитать пробеги ионов, но и позволит смоделировать какие образовались радиационные дефекты, как располагаются они в мишени и как они получились.
Материал и методы
Ионная имплантация – это процесс введения одного вещества (примеси) в другое вещество, из которого состоит мишень, при помощи бомбардировки ее поверхности ускоренными ионами. При бомбардировке поверхности мишени ускоренными частицами в результате множественных столкновений атомов мишени с налетающими атомами примеси происходит выбивание атомов из узлов решетки и возникают радиационные дефекты и изменяются свойства материала.
Большинство методов исследования процессов ионной имплантации являются деструктивными по отношению к мишени, так как для исследования получившихся слоев с примесью и дефектами структуры необходимо мишень послойно разрушить. Компьютерное моделирование позволит избежать разрушения.
Одним из перспективных методов моделирования является моделирование методом молекулярной динамики. Основным преимуществом которого является способность получить «реальную» микроскопическую динамику, подчиняющуюся ландшафту свободной энергии системы и межатомным взаимодействиям. В методе молекулярной динамики используются классические законы движения для расчета координат и момента частиц.
Для численного интегрирования динамических уравнений используются схемы конечно-разностной аппроксимации. Широко используется скоростной алгоритм Верле второго порядка точности. [1]
В данной работе рассматривается компьютерное моделирования процесса ионной имплантации с использованием пакета Lammps (http://lammps. sandia.gov). Приводится алгоритм настройки среды и подготовки входного файла для проведения моделирования. Проводиться анализ наличия точечных дефектов кристаллической структуры для комбинации ион-мишень: Cu-Fe с использованием алгоритма основанного на использовании ячеек Вигнера-Зейтца. Приводятся графики профиля распределения примеси и распределения точеных дефектов по глубине мишени. Для описания межатомных взаимодействий выбран потенциал EAM . Моделирование проводилось для материала идеальной структуры.
В таблице 1 приведены условия, для которых проводилось компьютерное моделирование и строились файлы входных данных.
Таблица 1
Условия эксперимента
|
Ион-мишень: Сu-Fe |
|
|
Энергии налетающих атомов, keV |
|
|
5 |
7 |
|
Размер ячейки моделирования, Å: 85.98x85.98x1433.0 |
|
|
Число итераций |
|
|
8500 |
10500 |
|
Число налетающих атомов: 2000 |
|
|
Температура, K: 300 |
|
|
Скорости налетающих атомов, Å/picosecond |
|
|
430 |
508,7 |
|
Файл межатомного потенциала [2]: FeCuNi.eam.alloy |
|
Методика проведения компьютерного моделирования процесса ионной имплантации заключается в следующем:
- выбор платформы для проведения моделирования;
- установка программного обеспечения;
- сборка программного пакета;
- создание файла со входными данными;
- запуск моделирования;
- анализ результатов выгрузки;
Выбор платформы для проведения моделирования методом молекулярной динамики необходимо проводить с учетом объемов вычислений и простоту в распараллеливании. Удачным вариантом будет использование сервера с графическими картами с технологией CUDA/openCL, либо кластера из нескольких (более двух) машин для проведения моделирования с использованием технологии MPI. Кроме того, на машинах должно быть достаточное количество оперативной памяти, в которой будут храниться данные о частицах системы. Для моделирования в рамках данной работы использовался MacBook Pro на macOS Sierra 2,5 GHz Intel Core i7 16 ГБ 1600 MHz DDR3. Вычисления проводились с использованием технологии MPI. Мощностей машины оказалось достаточно для моделирования сравнительно не больших мишеней (размер мишени по длине, ширине и высоте всего по несколько ангстрем, общие число атомов в системе порядка четырех миллионов частиц).
Установка программного обеспечения для проведения моделирования включает в себя установку пакета программ для классической молекулярной динамики LAMMPS [4] подходящей версии (использовалась версия от 31.03. 2017), установку библиотеки Vorono++ [3], для проведения анализа структуры материала, установку дополнительных системных библиотек, установку визуализатора и анализатора данных для атомистической симуляции.
Сборка программного пакета LAMMPS под конкретную платформу заключается в формировании make-файла и сборке. В самом LAMMPS уже есть стандартные готовые make-файлы (находящиеся в src/MAKE/MASHINES) для разных типов систем и имеющегося оборудования, но в каждом конкретном случае могут потребоваться дополнительные изменения со стороны пользователя. После составления make-файла необходимо включить пакеты, которые понадобятся при моделировании в сборку. Если необходимо использовать технологии CUDA/openCL и MPI, то для них придется скорректировать и запустить дополнительные make-файлы, прежде чем собрать сам LAMMPS. Пакеты могут быть стандартными, созданными и поддерживаемыми разработчиками LAMMPS, и пользовательскими, созданными пользователями пакета молекулярной динамики. Между пакетами могут быть зависимости, что стоит учитывать при сборке. Однако, не существует зависимости никаких стандартных пакетов от пользовательских. Как следствие, можно создать сборку только со стандартными пакетами и, в зависимости от задачи, использовать или не использовать пользовательские. Чтобы включить пакет в сборку, необходимо выполнить make yes-[имя пакета], чтобы исключить соответственно make no-[имя пакета],. Можно включить/выключить все пакеты разом: make yes/no-all. Статус пакетов (добавлен в сборку или нет) можно посмотреть командой make package-status. В данной работе для проведения моделирования использовались следующие пакеты: стандартные – ASPHERE, BODY, CLASS2, COLLOID, COMPRESS, CORESHELL, DIPOLE, GRANULAR, MISC, MOLECULE, MPIIO, OPT, PERI, PYTHON, QEQ, REPLICA, RIGID, SHOCK, SNAP, SRD, VORONOI. Пользовательские пакеты не использовались. Особо стоит отметить пакет VORONOI, необходимый для проведение анализа кристаллической структуры с использованием диаграммы Воронова. Для его корректной работы необходимо скачать библиотеку Vorono++ в виде архива с официального сайта распаковать его, собрать библиотеку с помощью make внутри распакованной директории (voro++-0.4.6), положить всю директорию с собранной библиотекой в /lib/voronoi и сделать здесь же две мягкие ссылки следующими командами ln -s voro++-0.4.6/src includelink и ln -s voro++-0.4.6/src liblink. После этого можно включать пакет VORONOI в сборку и собирать с ним LAMMPS. Для сборки с MPI необходимо перед сборкой LAMPPS собрать MPI пакет с помощью make mpi, затем make [имя машины]. Для данного моделирования команда выглядела следующим образом make –j 2 mac_mpi. Дополнительно, для сборки потребовалась установка интегрированной среды разработки программного обеспечения Xcode и библиотеки FFTW.
Запуск осуществляется командой mpirun -np 4 ~/local/bin/lmp_mac_mpi < implantation.impl, где mpirun -np 4 говорит, что необходимо использовать MPI с четырьмя процессами, а ~/local/bin/lmp_mac_mpi < implantation.impl указывает на путь к исполняемому файлу LAMMPS, полученному после сборки и путь к файлу со входными данными. Для macOS необходимо дополнительно перед запуском выполнить export TMPDIR=/tmp, чтобы сделать имя TMPDIR короче.
Структура файла входных данных устроена следующим образом:
- команды очистки формата и установка размерности области моделирования (2D или 3D);
- команда установки типа частиц, отражающего единицы всех величин, указанных во входном скрипте и файле данных, используется в самом начале входного скрипта, и может принимать одно из нескольких значений - lj, real, metal, si, cgs, electron, micro, nano;
- команды установки способа расчета третьего закона Ньютона для парных и валентных взаимодействий между атомами, либо независимо на каждом используемом процессоре, либо с обменом данными между ними, т. е. если на несколько процессоров попала одна и та же частица, то в первом случае расчет для нее будет проводиться независимо на каждом из них, а во втором - только на одном, а затем данные отправятся на остальные процессоры, где есть такая же частица;
- команды установки типа границ области моделирования, в частности, установка их периодичности (если граница периодическая, то вылетающий из нее атом появляется с противоположной стороны ячейки, если атом вылетает за непериодическую границу – он удаляется из области моделирования);
- команды установки типа атомов, предполагающие определение атрибутов связанных с атомами;
- команды установки определения структуры решетки для мишени;
- команды создания мишени и начального расположения атомов примеси:
- команды определение межатомного потенциала взаимодействия;
- команды задания частоты перестроения списков соседей;
- команды задания скоростей и фиксов (compute, fix), производящих перерасчет заданных характеристик для определенной группы атомов таких как, например, вычисление кинетической энергии, построение диаграммы Воронова для кристаллической решетки;
- команды определяющие параметры выгрузки данных о моделируемых частицах;
Для проведения последующего анализа количества дефектов и распределения примеси по глубине мишени проводилась выгрузка координат (x,y,z) атомов на определенных шагах вычислений, а также проводился дополнительный расчет наличия вакансий и включений с использованием диаграммы Воронова (построения решетки Вигнера-Зейтца, так как случай трехмерный).
Производилась выгрузка четырех типов. Первые три типа имеют сходный формат. Он включает общее число частиц (одного типа, если это выгрузка одного типа атомов или всех типов атомов, участвовавших в имплантации), общий коэффициент масштабирования, три вектора глобальной ячейки моделирования, массу атома, его химический символ, основные и дополнительные параметры (координаты, масштабированные по размеру ячейки, энергию, идентификаторы и т.п.).
Четвертый тип выгрузки предназначен для данных о результатах построения разбиения Воронова и анализа количества атомов в каждой получившейся области. Результат представлен в виде таблицы из трех колонок. В первой колонке идентификатор атома, во второй колонке количество атомов в ячейке, полученной при начальном разбиении, на нулевом шаге моделирования, в текущий момент времени, в третьей колонке количество атомов в ячейке, в ее настоящем положении в текущий момент времени.
Анализ выгрузки и построение графиков после завершения моделирования проводился с помощью скрипта на языке программирования Python.
Первой задачей был анализ распределения атомов по глубине мишени в зависимости от энергии, с которой они бомбардируют мишень. Для этого использовались файлы выгрузки данных об атомах на конечном этапе моделирования.
Скрипт получает на вход dump-файл для имплантируемого вещества, проходит по нему и составляет структуру-словарь, ключами которого являются идентификатор атома, а значением – структура-словарь с масштабированными по размеру области моделирования координатами x,y,z. Следующим шагом производиться преобразование координат в ангсремы (Å) и вычисление расстояние, пройденного от поверхности мишени (глубины проникновения) путем вычитания координаты z из размера мишени. Затем, вся мишень разбивается на слои толщиной по несколько ангрстрем и для каждого атома определяется в каком слое он оказался на основании вычисленной глубины проникновения в мишень. После этого строится график с использование библиотеки matplotlib.
Второй задачей было построение распределения точечных дефектов, вакансий и включений, по глубине мишени. Для этого использовались dump-файлы для материала мишени на начальном, нулевом, и конечном шаге моделирования. Нахождение вакансий и включений, данные о которых содержаться в dump-файле происходит с помощью построения решетки Вигнера-Зейтца. В пакете LAMMPS это реализует “compute voronoi/atom occupation”.
Скрипт считывает данные из dump-файла с дефектами, составляются два списка идентификаторов атомов, отдельно для вакансий и включений. Для вакансий, выбираются идентификаторы атомов у которых в первом столбце стоял 0. Для включений, выбираются идентификаторы атомов у которых значение во втором столбце было строго больше 1. Для атомов из начального и конечного dump-файла составляется структуры-словари, содержащие в качестве ключей идентификаторы атомов, в качестве значений - структуру-словарь с масштабированными по размеру области моделирования координатами x,y,z. Происходит преобразование координат в ангсремы (Å). Затем, для вакансий, из атомов начальной конфигурации выбираются только те, чей идентификатор есть в списке идентификаторов вакансий, для включений из конечной конфигурации выбираются те атомы, идентификаторы которых есть в списке включений. Мишень разбивается на слои толщиной по несколько ангстрем, для всех отобранных атомов определяется их принадлежность к определенному слою мишени и строятся графики распределения вакансий и включений по глубине мишени.
Результаты и обсуждение
Таблица 2
Количество вакансий и включений в мишени из железа после проведения ионной имплантации
|
|
Сu-Fe |
|
|
5keV |
7keV |
|
|
вакансии |
1646 |
810 |
|
включения |
5775 |
3800 |
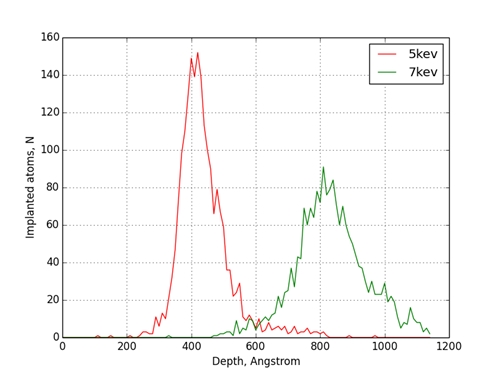
Рис.1 Распределение атомов меди по глубине мишени из железа
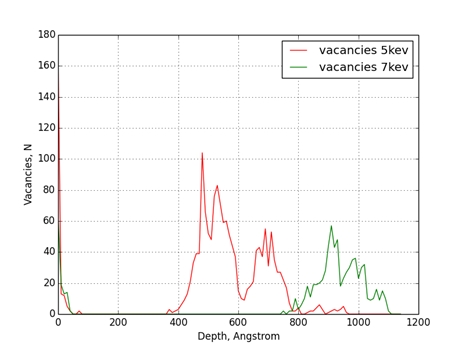
Рис. 2 Распределение вакансий по глубине мишени из железа
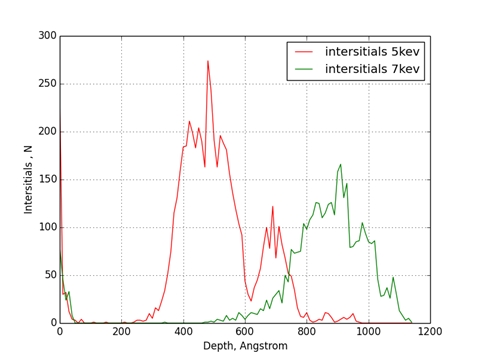
Рис. 3 Распределение включений по глубине мишени из железа
Выводы
Было проведено моделирование процесса ионной имплантации для комбинации ион-мишень Cu-Fe. Энергии имплантируемых атомов меди были равны 5keV и 7keV. В результате моделирования отмечено, что при увеличении энергии бомбардирующих атомов максимум концентрации примеси смещается вглубь мишени и становиться более растянутым. Максимумы концентрации вакансий и включений при увеличении энергии бомбардирующих атомов также смещаются вглубь мишени. Стоит отметить, что они находятся глубже чем, максимумы концентрации примеси в обоих случаях.
Чтобы провести компьютерное моделирование процесса ионной имплантации с помощью пакета для классической молекулярной динамики LAMMPS необходимо выбрать оборудование исходя из предполагаемых объемов задачи, настроить программное обеспечение и сформировать входной фай с учетом участвующих в моделировании элементов. Для анализа результатов может потребоваться дополнительно написание кода.
1. Углов В.В. Радиационные процессы и явления в твердых телах: учеб. пособие / В.В. Углов. – Минск: Вышэйшая школа, 2016. – 188с.
2. http://www.ctcms.nist.gov/potentials - Interatomic Potentials Repository Project (дата последнего обращения 10.06.2017)
3. http://math.lbl.gov/voro++/ - Voro++ (дата последнего обращения 10.06.2017)
4. http://lammps.sandia.gov/ - LAMMPS Molecular Dynamics Simulator (дата последнего обращения 10.06.2017)
5. http://geant4.cern.ch/ - GEANT 4 (дата последнего обращения 10.06.2017)
6. http://www.srim.org/ - SRIM (дата последнего обращения 10.06.2017)
7. http://www.physics-in-a-nutshell.com/article/11 - Physics in a nutshell (дата последнего обращения 7.05.2017)
8. L. Himics, S. Tóth, M. Veres, A. Tóth, M. Koós Effective implantation of light emitting centers by plasma immersion ion implantation and focused ion beam methods into nanosized diamond // Applied Surface Science, № 328, 2015, - C577–582.
9. Lisa Gamer, Christoph E. Düllmann, Christian Enss, Andreas Fleischmann, Loredana Gastaldo, Clemens Hassel, Sebastian Kempf, Tom Kieck, Klaus Wend Simulation and optimization of the implantation of holmium atoms into metallic magnetic microcalorimeters for neutrino mass determination experiments // Nuclear Instruments and Methods in Physics Research A, № 854, 2017, - C139 -148.
10. S.K. Sharma, P.K. Pujari Embedded Si nanoclusters in a-alumina synthesized by ion implantation: An investigation using depth dependent Doppler broadening spectroscopy // Journal of Alloys and Compounds, № 715, 2017, - C247 – 253.
11. Julien Bordes, Sébastien Incerti, Nathanael Lampe, Manuel Bardiès, Marie-Claude Bordage Low-energy electron dose-point kernel simulations using new physics models implemented in Geant4-DNA // Nuclear Instruments and Methods in Physics Research B, №398, 2017, - C13–20.
12. А.Ю.Куксин, А.В. Янилкин Образование дефектов в каскадах смещений в молибдене: моделирование методом молекулярной динамики // Физика металлов и металловедение, 2016, Т. 117, № 3, - с. 230-237.
13. Е.С. Парфенова Математическое моделирование начальной̆ стадии процесса ионной̆ имплантации // Известия высших учебных заведений. Физика, 2015, Т. 58, № 6/2, - С. 226-229.
14. Окунев И.В. Единая теория гравитации и электромагнетизма //СтройМного, 2016. №2 (3). URL: http://stroymnogo.com/science/economy/edinaya-teoriya-gravitatsii-i-elekt/
15. Philipp Neumann, Hanno Flohr, Rahul Arora, Piet Jarmatz, Nikola Tchipev, Hans-Joachim Bungartz MaMiCo: Software design for parallel molecular-continuum flow simulations // Computer Physics Communications, № 200, 2016, - С324–335.
16. L. Pentecoste, P. Brault, A.-L. Thomann, P. Desgardin, T. Lecas, T. Belhabib, M.-F. Barthe, T. Sauvage Low energy and low fluence helium implantations in tungsten: Molecular dynamics simulations and experiments // Journal of Nuclear Materials, № 470, 2016, - С44-54.
17. G. S. Smirnov, V. V. Stegailov Efficiency of Classical Molecular Dynamics Algorithms on Supercomputers // Mathematical Models and Computer Simulations, 2016, Т. 8, № 6, -С734–743.
18. Evangelos Voyiatzis, Michael C. Böhm, Florian Müller-Plathe A geometric approach to identify cavities in particle systems // Computer Physics Communications, №196, 2015, -С247–254.
19. Changwoo Kang, Qingyu Wang , Lin Shao Kinetics of interstitial defects in a-Fe: The effect from uniaxial stress // Journal of Nuclear Materials, №485, 2017, - С159-168.
20. A.V. Korchuganov, K.P. Zolnikov, D.S. Kryzhevich, V.M. Chernov, S.G. Psakhie Generation of shock waves in iron under irradiation // Nuclear Instruments and Methods in Physics Research B, №352, 2015, -С39–42.
21. Pedro López, Iván Santos, María Aboy, Luis A. Marqués, Lourdes Pelaz A detailed approach for the classification and statistical analysis of irradiation induced defects // Nuclear Instruments and Methods in Physics Research B, №352, 2015, -С156–159.
1. Uglov V.V. Radiatsionnyye protsessy i yavleniya v tverdykh telakh: ucheb. Posobiye V.V. Uglov. Minsk: Vysheyshaya shkola, 2016. 188p.
2. http://www.ctcms.nist.gov/potentials - Interatomic Potentials Repository Project (last access date10.06.2017)
3. http://math.lbl.gov/voro++/ - Voro++ (last access date10.06.2017)
4. http://lammps.sandia.gov/ - LAMMPS Molecular Dynamics Simulator (last access date 10.06.2017)
5. http://geant4.cern.ch/ - GEANT 4 (last access date10.06.2017)
6. http://www.srim.org/ - SRIM (last access date10.06.2017)
7. http://www.physics-in-a-nutshell.com/article/11 - Physics in a nutshell last access date 7.05.2017)
8. L. Himics, S. Tóth, M. Veres, A. Tóth, M. Koós Effective implantation of light emitting centers by plasma immersion ion implantation and focused ion beam methods into nanosized diamond in Applied Surface Science, No 328, 2015, Pp. 577–582.
9. Lisa Gamer, Christoph E. Düllmann, Christian Enss, Andreas Fleischmann, Loredana Gastaldo, Clemens Hassel, Sebastian Kempf, Tom Kieck, Klaus Wend Simulation and optimization of the implantation of holmium atoms into metallic magnetic microcalorimeters for neutrino mass determination experiments in Nuclear Instruments and Methods in Physics Research A, No 854, 2017, Pp. 139 -148.
10. S.K. Sharma, P.K. Pujari Embedded Si nanoclusters in a-alumina synthesized by ion implantation: An investigation using depth dependent Doppler broadening spectroscopy in Journal of Alloys and Compounds, No 715, 2017, Pp. 247 – 253.
11. Julien Bordes, Sébastien Incerti, Nathanael Lampe, Manuel Bardiès, Marie-Claude Bordage Low-energy electron dose-point kernel simulations using new physics models implemented in Geant4-DNA in Nuclear Instruments and Methods in Physics Research B, No 398, 2017, Pp. 13–20.
12. A.Y. Kuksin, A.V. Ynilkin Formation of Defects in Displacement Cascades in Molybdenum: Simulation of Molecular Dynamics in The Physics of Metals and Metallography, 2016, Vol. 117, No 3, Pp. 230-237.
13. E.S. Parfenova Mathematical modeling of ion implantation process in Russian Physics Journal, 2015, Vol. 58, No 6/2, Pp. 226-229.
14. Okunev I.V. Edinaja teorija gravitacii i jelektromagnetizma.StrojMnogo, 2016. No 2 (3). URL: http: stroymnogo.com/science/economy/edinaya-teoriya-gravitatsii-i-elekt/
15. Philipp Neumann, Hanno Flohr, Rahul Arora, Piet Jarmatz, Nikola Tchipev, Hans-Joachim Bungartz MaMiCo: Software design for parallel molecular-continuum flow simulations in Computer Physics Communications, No 200, 2016, Pp. 324–335.
16. L. Pentecoste, P. Brault, A.-L. Thomann, P. Desgardin, T. Lecas, T. Belhabib, M.-F. Barthe, T. Sauvage Low energy and low fluence helium implantations in tungsten: Molecular dynamics simulations and experiments in Journal of Nuclear Materials, No 470, 2016, Pp. 44-54.
17 G. S. Smirnov, V. V. Stegailov Efficiency of Classical Molecular Dynamics Algorithms on Supercomputers in Mathematical Models and Computer Simulations, 2016, Vol. 8, No 6, Pp. 734–743.
18. Evangelos Voyiatzis, Michael C. Böhm, Florian Müller-Plathe A geometric approach to identify cavities in particle systems in Computer Physics Communications, No 196, 2015, Pp. 247–254.
19. Changwoo Kang, Qingyu Wang , Lin Shao Kinetics of interstitial defects in a-Fe: The effect from uniaxial stress in Journal of Nuclear Materials, No 485, 2017, Pp. 159-168.
20. A.V. Korchuganov, K.P. Zolnikov, D.S. Kryzhevich, V.M. Chernov, S.G. Psakhie Generation of shock waves in iron under irradiation in Nuclear Instruments and Methods in Physics Research B, No 352, 2015, Pp. 39–42.
21. Pedro López, Iván Santos, María Aboy, Luis A. Marqués, Lourdes Pelaz A detailed approach for the classification and statistical analysis of irradiation induced defects in Nuclear Instruments and Methods in Physics Research B, No 352, 2015, Pp. 156–159.
архив: 2013 2012 2011 1999-2011 новости ИТ гость портала 2013 тема недели 2013 поздравления
E-mail редакции: vzh85@yandex.ru,